|
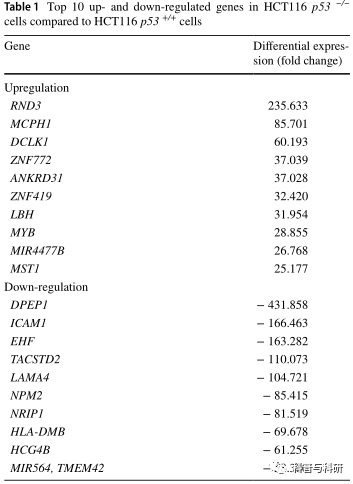
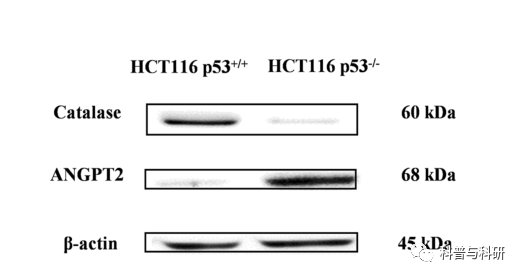
第83期:p53缺失的结肠癌细胞基因组和转录组分析鉴定潜在的新耐药机制 摘要 TP53在肿瘤抑制中起关键作用,50%的侵袭性肿瘤显示TP53基因突变。在本研究中,我们描述了具有TP53缺失的结肠癌细胞(HCT116 p53−/−),TP53缺失是来源于HCT116-p53+/+细胞的一个亚系。进行RNA测序和网络分析以确定新的耐药机制。用多色荧光原位杂交(mFISH)和阵列比较基因组杂交(aCGH)鉴定染色体畸变。许多基因在HCT116 p53−/−细胞中过度表达:RND3/RhoE(235.6倍上调)、DCLK1(60.2倍上调)、LBH(31.9倍上调)、MYB(28.9倍上调)、TACSTD2(110.1倍下调),根据先前发表的研究,NRIP1(81.5倍下调)和HLA-DMB(69.7倍下调)是已鉴定的对多药耐药(MDR)有潜在影响的基因,它们与癌症进展和肿瘤发生有关。可能是由于TP53缺失,DNA修复和凋亡的紊乱导致了细胞和机体组织的异常,最终增加了肿瘤的发生和发展潜力。NFκB、PI3K和HSP70是融合蛋白网络的中心,TH1-2通路是受影响的通路之一,可以推测炎症通路与细胞周期调控和热休克反应共同参与耐药表型。HCT116-p53−细胞比HCT116-p53+/+细胞有更多的染色体畸变、拷贝数的增加和减少。综上所述,许多基因组畸变可能与未知的耐药机制有关。这可能对未来的治疗策略有重要的影响。 简介 TP53被描述为基因组的守护者。当外来和致癌物质造成有害损害时,p53保持细胞完整性。DNA的畸变和损伤被p53识别,导致细胞周期阻滞和DNA修复。当持续性损伤超过细胞修复能力极限时,p53可触发细胞凋亡。p53诱导凋亡的机制包括FAS、KILLER/DR5和线粒体途径的转录激活。此外,细胞存活,如BCL2,IGFR、MCL-1、survivin和PIK3CA被p53抑制。P53在翻译后修饰(如磷酸化、乙酰化和甲基化)激活后,除了凋亡和生长停滞外,还参与各种下游过程。作为一种转录因子,通过调节细胞周期阻滞、DNA修复和/或凋亡相关途径维持基因组完整性。在细胞周期调控方面,p53激活p21/waf1,p21/waf1是G2/M特异性细胞分裂控制蛋白2激酶和细胞周期蛋白依赖性G1激酶的抑制剂,随后导致G2/M和G1检查点控制。由于突变的p53而不能在G1和G2/M检查点阻止细胞可导致耐药性。 DNA修复和凋亡机制对于维持人体细胞的健康状态至关重要。在正常情况下,过度DNA损伤或其他畸变的细胞通过凋亡被消除。如果p53启动的这种调控机制被破坏,就会出现异常的细胞增殖和过度的DNA损伤。p53基因突变是DNA修复和凋亡中断的主要原因之一,而DNA修复和凋亡可能是由于异常细胞数量的增加而引发的,异常细胞更容易发生突变和染色体不稳定。由于无功能的p53而导致DNA损伤累积的细胞的异常增殖也会影响具有额外突变的下一代细胞。最终,这会增加致癌的风险。 超过50%的人类癌症发生P53突变,而结直肠癌是经常发生有害P53突变的癌症类型之一。大多数突变发生在DNA结合域,并导致蛋白质错误折叠或破坏DNA结合能力。凋亡功能的丧失是放射和耐药癌细胞发展的一个重要原因。此外,具有p53突变的肿瘤通常具有加重转移和基因组不稳定性的特征。突变型p53的其他致癌功能包括促进侵袭、迁移、血管生成和增殖,从而导致耐药性增强和有丝分裂缺陷。上述功能只是突变型p53调控癌症进展的众多途径中的一小部分。例如,p53还通过限制糖酵解和促进线粒体呼吸对药物代谢和细胞代谢产生影响。 在介导多药耐药(MDR)表型的ATP结合盒(ABC)转运蛋白中,对多种药物的耐药性已经得到了广泛的研究。然而,多重耐药性并不局限于ABC转运体,其他多药耐药现象也已被描述,包括p53、Bcl-2、肿瘤增殖率等。先前对HCT116细胞系进行了微阵列分析,但是基因组学和转录组学方法在同基因敲除细胞中的应用使得细胞系之间能够进行更好和更深入的比较,以确定新的耐药机制。 在这项研究中,我们应用RNA测序,阵列比较基因组杂交(aCGH)和多色荧光原位杂交(mFISH)分析HCT116p53+/+结肠癌细胞及其p53缺失的耐药亚系HCT116 p53−/−来表征基因,通路,蛋白质网络和染色体畸变与HCT116 p53−/−细胞系的耐药性有关。总的来说,这项研究将提供一个完整的复杂的机制和遗传改变结肠癌细胞及其贡献的耐药发生在p53缺失的概述。 材料与方法 细胞培养 HCT116 p53+/+及其耐药HCT116 p53−/−亚系。HCT116 p53−/−细胞具有显著的有丝分裂检查点缺陷,因此它们不能正常对DNA损伤剂作出反应,进入有丝分裂,随后在DNA损伤的情况下复制其基因组。在过去的几年中,已经研究了HCT116 p53−的耐药谱。与野生型细胞相比,这些敲除细胞对已建立的多种药理类别的抗癌药物(阿霉素、5-氟尿嘧啶和5′-脱氧-5-氟尿嘧啶、顺铂和奥沙利铂、足叶乙甙、,以及研究具有抗癌活性的细胞毒性化合物(三氧化二砷作为PML/RARA抑制剂,nutlin-3a作为p53激活剂,氟嘧啶F10,HDAC抑制剂entinostat和合成多胺DENSpm)和甚至细胞毒性但非癌症药物(抗疟药奎那克林,抗惊厥药)丙戊酸钠与布洛芬的抗炎及COX1/2抑制作用 RNA测序 基因表达采用FPKM(每千碱基转录物每百万映射读取片段数)测量进行量化(Choudhri et al.2018;Wesolowski et al.2013)。HCT116 p53−/−细胞中基因的解除调控是通过将HCT116 p53−/−细胞中基因的总FPKM值除以HCT116p53+/+细胞中的FPKM值来计算的。 路径与网络分析 应用±7的RNA表达的倍数变化进行过滤,然后对解除调控的基因列表进行Ingenuity Pathway Analysis(IPA)(QIAGENRedwood City,USA,www.qiage.comn、 com/Ingenuity)识别HCT116 p53−细胞中的特定网络和通路。 荧光分析系统 HCT116 p53−/−和HCT116 p53+/+细胞根据标准程序进行细胞遗传学制备以获得中期扩散,并使用分子细胞遗传学进行分析。如先前报道的,使用人类全染色体涂料作为探针进行mFISH。 基因体检查 使用QIAmp DNA迷你试剂盒,从HCT116p53−/−和HCT116 p53+/+细胞中提取全基因组DNA。aCGH按照之前的报告进行。 蛋白质印迹 在HCT116 p53−/−和HCT116 p53+/+细胞中评估所选基因(即ANGPT2和过氧化氢酶)的蛋白质表达水平,以验证通过RNA测序分析发现的调控解除。用蛋白提取缓冲液(M-PER)从细胞中提取总蛋白™ 哺乳动物蛋白提取试剂与1%Halt混合™ 蛋白酶抑制剂鸡尾酒。将相当于30μg的样品加载到10%SDS-PAGE中进行分离,然后转移到Ruti®-PVDF膜中。用5%BSA封闭膜1小时,并在4°C下用选定的抗ANGPT2、过氧化氢酶和β-肌动蛋白的一级抗体进行探测(1:1000)。24h后,用HRP二级抗体(1:2000)孵育1h,用Luminata检测™ Classico Western HRP基板。图像分析采用ImageJ软件。 结果 HCT116 p53−细胞的差异基因表达谱、下游途径和网络分析 与HCT116 p53+/+细胞相比,HCT116 p53-细胞中每个基因表达的RNA-seq衍生FPKM值的比率被认为是基因表达的倍数变化。为了进一步分析,考虑了阈值为±7的差异基因表达,得出了300个差异表达基因。表1列出了HCT116-p53-细胞中前10个上调和下调基因。RND3(+235.6)、MCPH1(+85.7)和MYB(+28.9)是上调最多的基因,而DPEP1(−431.9)、ICAM1(−166.5)和NPM2(−85.4)是下调最多的基因。 表1与HCT116 p53 +/+细胞相比,HCT 116 p53/+细胞中前10个上调和下调基因 在网络1中,组蛋白H4、细胞周期蛋白A和NFκB的节点数最多,CD3和Hsp70的节点数最多。网络1中的“癌症”、“组织损伤和异常”,而网络2中的“细胞组装和组织”和“分子运输”是受影响的生物学功能。Erk1/2在网络3中的节点数最高。“机体损伤和异常”和“碳水化合物代谢”是网络3中受影响的生物功能(图1)。 图1与HCT116p53 +/+细胞相比,HCT 116 p53/-细胞中受影响的蛋白质网络。用绿色标记的基因被下调,用红色标记的基因被上调。 一些已知参与耐药的基因被解除调控,这意味着HCT116 p53−细胞表现出多因子耐药表型。如果应用±7.0的倍数变化阈值,则一个DNA修复基因、一个氧化应激基因和一个转录因子基因属于解除调控的耐药基因,这意味着这些基因类别的基因可能对HCT116 p53−的MDR表型有重要影响。这些基因如表2所示,所有参与抗性机制的解除调控基因的完整列表如补充表2所示。 表2与野生型HCT116 p53 +/+细胞相比,HCT 116 p53/+细胞中参与经典耐药机制的去调节基因(阈值:表达改变7倍) 对前三大网络进行了合并,并对合并后的网络进行了进一步分析。(图2)所示,NFκB与PI3K和HSP70一起位于融合网络的中心。 图2网络1、2和3的合并蛋白质网络 “癌症”、“机体损伤和异常”以及“细胞间信号和相互作用”是HCT116p53−细胞中受影响最大的生物学功能(图3)。位于前10个生物功能列表中的基因如补充表3所示。 图3与HCT116 p53 +/+细胞相比,HCT116 p53 +/细胞中受影响的生物学功能(前10)。橙色线表示统计显著性阈值(p = 0.05) “Th1通路”(p值:0.000437)、“IL4信号通路”(p值:0.012589)是HCT116 p53−细胞中最重要的信号通路之一,这意味着可能的免疫应答通路对耐药性产生影响(图4)。 图4与HCT116 p53 +/+细胞相比,HCT116 p53 +/细胞中受影响的信号通路(前10)。橙色线描绘了统计显著性阈值(p = 0.05),橙色图描绘了每个途径中去调控基因的比例 在116个p53靶基因中(Fischer 2017),33个下调,19个上调(倍数变化高于±1.5的阈值),如表3所示。 表3与HCT116-p53 +/+细胞相比,HCT 116 p53/-细胞中p53靶基因的去调节 在蛋白质水平上对所选基因进行ANGPT2和过氧化氢酶的验证。如图5所示,在HCT116 p53−细胞中,ANGPT2上调(+8.7倍),而过氧化氢酶下调(−1.9倍),与RNA测序输出相关,并在蛋白质水平验证RNA表达数据。 图5通过蛋白质印迹法测定的血管生成素样蛋白2和过氧化氢酶在HCT116 p53和HCT116p53 +/+细胞中的表达 mFISH HCT116-p53 +/+细胞显示核型45 < 2n >,X,dup(10)(q?q?),der(16)t(8;16)(p13;?),der(18)t(17;18) (?q;p11.2),而HCT 116-p53/-细胞具有45 < 2n > X,t(5;7)(q1?3;p22),dup(10)(q?q?),der(16)t(8;16)(p13;?),der(18)t(17;18)(?q;p11.2)。图6中描述了微荧光分析的结果。HCT116-p53细胞具有克隆性从头平衡易位t(5;7)(q1?3;p22)与HCT116-p53 +/+细胞相比。 图6:HCT116 p53+/+(a)和HCT116p53−/—(b)细胞的mFISH分析 aCGH HCT116 p53 +/+:染色体扩增和缺失在RNA Seq分析中观察到的基因表达的去调控中得到很好的反映。LVRN下调3.3倍,AP3S1上调1.7倍。相应的染色体基因座(5q23.1)被扩增。FLJ42393下调了1.7倍。在相应的染色体位点(3q 27.3–q28)有一个缺失。结果总结在表4中。HCT116 p53/:与HCT116 p53 +/+细胞相比,在HCT 116-p53/+细胞中观察到更多的扩增和缺失。这意味着p53缺失导致额外的染色体畸变、扩增和缺失的积累。NXPH2上调了1.7倍,并且在相应的染色体座位上有扩增。OR5K2上调了1.7倍,在相应的染色体座位上有扩增。PARK2下调了8.1倍,在相应的染色体位点上有一个缺失。aCGH数据与RNA-Seq结果的相关性清楚地显示了基因在相应染色体基因座扩增/缺失的差异表达,如表4所示。表5描述了总体aCGH结果。 表4染色体畸变和相应的去调控基因。aCGH和核糖核酸测序图谱的比较 表5:aCGH总体结果 讨论 在本研究中以结肠癌细胞系HCT116 p53+/+和具有TP53缺失的耐药HCT116p53-亚系为模型,旨在鉴定新的耐药基因。通过RNA测序、mFISH和aCGH鉴定基因表达谱、信号通路、生物学功能和染色体异常,一些已知参与耐药的基因被解除调控,支持HCT116 p53−细胞的多因子耐药表型,这些基因在各种耐药簇中被鉴定,包括凋亡、DNA修复、铁下垂、谷胱甘肽相关、热休克、氧化应激,补充表2中列出的转录因子可能对MDR表型有重要影响。 最上调的基因RND3/RhoE(+235.6倍)以前与肿瘤的侵袭、转移有关,并被报道为肿瘤耐药的潜在标志物胃癌以及结直肠癌病例的复发和预后。CARD11突变与伊布替尼有关。CARD11(−7.4倍)出现在前五大生物学功能的基因列表中,表明细胞凋亡抑制下调CARD11可能在HCT116p53−细胞的耐药表型中起重要作用。据报道,CARD11对癌症中的伊布替尼耐药有贡献,即CARD11可能在HCT116p53−/−细胞的耐药表型中发挥作用。据报道,DCLK1(+60.2倍)与非小细胞肺癌细胞对顺铂的耐药相关,miR539靶向DCLK1导致对顺铂的敏感性增加。DCLK1也与结直肠癌、胰腺癌和肾癌的耐药性相关。据报道,LBH(+31.9倍)是肝细胞癌的潜在标志物,因为其过度表达与不良预后相关。Myb(+28.9倍表达于敲除细胞)是一种致癌转录因子,在促进白血病细胞转化中发挥作用。Myb与结肠癌细胞的顺铂耐药性有关。它还参与一些实体瘤的发展和进展,包括黑色素瘤TACSTD2的缺失通过抑制化疗药物诱导的细胞凋亡促进鳞癌的进展和耐药性,这意味着TACSTD2可以作为鳞状细胞癌病理分级的标志物(图1表2与野生型HCT116 p53+/+细胞相比,HCT116 p53-细胞中参与经典耐药机制的解除调控基因(阈值:±7倍变化表达)差异表达(倍变化)DNA修复XRCC211.200 BRIP1 7.711氧化应激NCF215.131 MB−8.093转录因子MYB 28.855 NFATC4−7.619 。在HCT116 p53−/−细胞中下调110.1倍,指出TACSTD2下调可能是导致HCT116 p53−/−细胞侵袭性生长和多药耐药的机制。NRIP1下调后,食管鳞状细胞癌细胞的迁移和侵袭能力增强。本文观察到NRIP1在HCT116p53−细胞中下调81.5倍,这意味着NRIP1下调可能在多药耐药表型中发挥作用。HLA-DMB(−69.7倍)属于主要的组织相容性复合体II类基因,晚期浆液性卵巢癌中,HLA-DMB的高表达通过增加CD8淋巴细胞数量与较高的生存率相关。HLADMB的下调可能通过影响肿瘤侵袭性与HCT116 p53−细胞的MDR表型有关。 MYB转录因子的过度表达(+28.8倍于敲除细胞的表达)与不良预后相关,并且经常在结直肠癌(CRC)中观察到。另一项研究指出,MYB在肿瘤细胞中的表达由于其致瘤作用而调节宿主免疫反应,这有可能影响CRC患者免疫治疗的使用。MYC(+5.1倍)是另一种在肿瘤发生中起关键作用的转录因子。它调节细胞周期相关基因的表达,并且在各种癌症类型中观察到过表达,包括结肠癌。 本实验已经在HCT116 p53−细胞中确定了最受影响的信号通路中的IL4信号通路,这意味着免疫应答通路可能影响耐药性。一项研究表明,IL-4可增强BCR信号传导,降低BCR激酶抑制剂(如伊布替尼)在CLL细胞中的有效性。另一项研究报告,通过白细胞介素-1受体相关激酶1和4(IRAK1/4)复合物激活先天免疫途径有助于FLT3突变AML细胞的适应性抵抗。这一结果支持我们观察到的免疫应答途径与耐药性的关系。 RNA-seq结果通过westernblotting对ANGPT2和过氧化氢酶进行验证。与HCT116 p53-+/+细胞相比,在HCT116 p53-/-细胞中,angpt2mrna上调,而CAT mRNA下调。蛋白表达证实了这一点。CAT在肿瘤中经常下调,例如,乳腺癌的特点是过氧化氢酶下调并伴随SOD过度表达。另一方面,ANGPT2的上调与结肠癌的肝转移有关。 网络分析指出,网络1的“癌症”、“机体损伤与异常”、“细胞组装与组织”、“分子运输”网络2的“机体损伤与异常”、“碳水化合物代谢”网络3为主要影响因素功能。由于TP53缺失,DNA修复和凋亡机制的破坏可能导致细胞和组织结构的异常,最终增加了肿瘤的发生和发展潜力。在网络1中,编码组蛋白H4、细胞周期蛋白A和NFκB的基因具有最高的结点数。CD3和HSP70在网络2的中心有最多的节点,这意味着细胞周期调控的影响炎症和热休克反应对耐药性的影响。重要的是,网络2中分子转运基因的出现强调了p53和细胞转运蛋白之间可能存在的相互作用,从而促进癌细胞的MDR。ABC转运蛋白的一个成员ABCB1/MDR1基因在转录上依赖于p53,其中野生型p53通过序列特异性结合下游启动子对ABCB1/MDR1基因表达产生负面影响。相反,突变型p53在不同细胞系中激活ABCB1/MDR1启动子。ERK1/2基因在网络3中具有最高的节点数,表明ERK调控的细胞增殖途径对耐药性的贡献。 NFκB与PI3K和HSP70一起位于融合网络的中心,这意味着炎症途径以及细胞周期和热休克反应在MDR表型中的作用。Th1、Th2通路和CD28信号通路是HCT116-p53-细胞中最受影响的信号通路之一,支持炎症通路在MDR表型中起重要作用的假说。 结论 总的来说,通过RNA测序、mFISH和aCGH分析结肠癌细胞系HCT116 p53和HCT116p53 +/+的基因表达谱,以比较的方式鉴定差异表达的基因、受影响的蛋白质网络、途径、生物学功能以及染色体畸变。发现了多种基因、途径和网络,它们可能与结肠癌的耐药性和侵袭性行为有关。这项研究清楚地表明,TP53敲除细胞的耐药性是由多个因素决定的,而不是由单个因素决定的。显然,多因素耐药性使新治疗策略的开发变得复杂。然而本研究可能代表了一个设计更具体和更有前途的绕过耐药性的抗癌策略的起点。 供稿:朵 朵 审核:戴西件 排版:静 静 |